
Gene editing therapy from CHOP treats ultra-rare metabolic disorder CPS1
On May 15 2025, at the annual meeting for the American Society for Gene and Cell Therapy, doctors from the Children’s Hospital of Pennsylvania (CHOP) presented the striking case report of KJ Muldoon, an infant who had been treated and potentially cured by gene editing technology (news summary here; original article here). At just one week old, KJ was diagnosed with CPS1 deficiency, an ultra-rare metabolic disorder similar to Barth syndrome that affects 1 in 1.3 million people and is associated with high mortality in infancy. With unprecedented speed, academic scientists, commercial partners, and federal regulators worked together to develop a custom gene editing treatment for KJ in just 6 months. Regulatory approval to test the therapy was given in <1 week and doctors were able to administer two doses of the therapy before KJ’s 8th month of life.
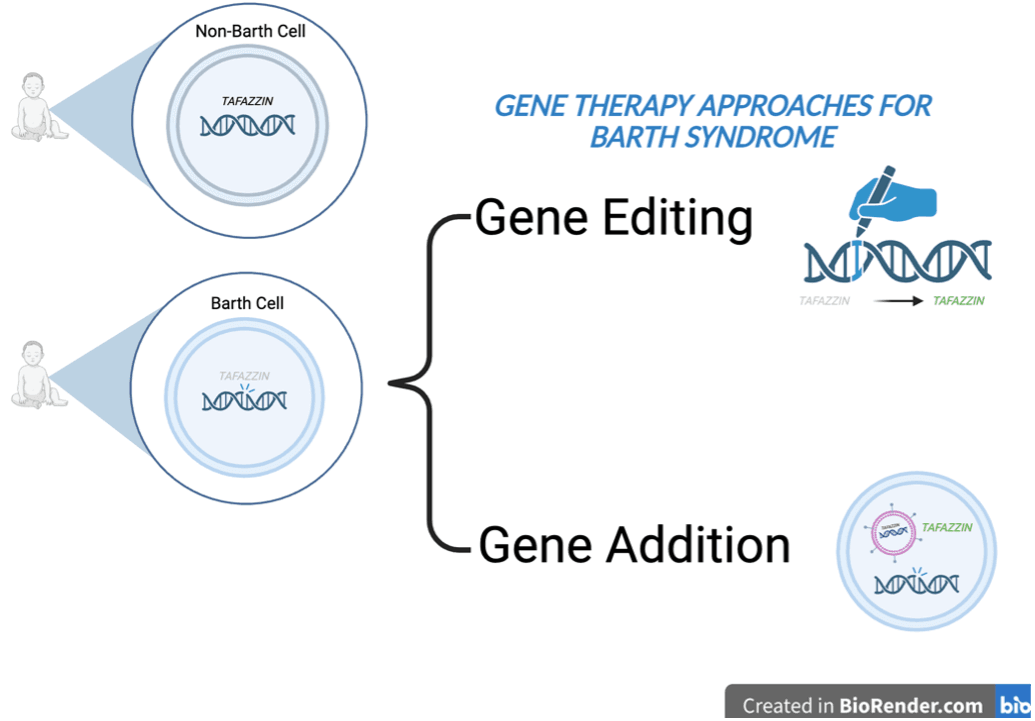
What is gene editing? What is gene addition?
Several different types of gene therapy have been developed for disorders like Barth syndrome where a gene is no longer working. In KJ’s case above, scientists developed a gene editing approach utilizing CRISPR, a technology bolstered by >10 years of academic and commercial research and topic of the 2020 Nobel Prize in Chemistry (read a lay-friendly primer about CRISPR here). Gene editing can be thought of like the “cut and paste” feature in Microsoft Word. In this therapy, a special tool called a guide RNA is designed to recognize a specific variant or typo of interest. The guide RNA looks through all of the DNA in each cell to find the typo and, in conjunction with a pair of molecular scissors called Cas9 and a corrected DNA template, the variant is removed and replaced with the corrected version. Prior to KJ’s story, two other CRISPR-based gene editing therapies have been approved by the FDA, but both are used on hematopoietic stem cells that get treated outside of the body and then returned.
Another form of gene therapy is called gene addition. Here, DNA containing a corrected version of the gene is enveloped in a virus that has been modified to find and infect cells but not make the recipient sick. The virus is then delivered via intravenous injection. Rather than fixing the variant in each cell, this therapy can be used to provide a corrected copy of the gene to each cell. This approach works best when a gene is no longer functional but does not work well in disorders where too much of a gene is produced. Barth Syndrome Foundation has funded gene therapy efforts from several labs and is currently supporting Dr. William Pu’s lab at Boston Children’s Hospital through the Strategic Initiatives Program.
Which technology is best for Barth syndrome?
Currently, there isn’t a simple answer to the question posed above. Gene addition and gene editing each have strengths and limitations in the context of Barth syndrome. Gene editing directly treats the underlying variant or mutation in the affected individual’s own cells, thus permanently correcting the issue. Gene addition relies on providing a corrected copy of the gene to each cell, but the underlying mutation continues to exist. In Barth syndrome, over 200 different DNA variants are known to cause the syndrome. While a single gene addition therapy can be given to a population that harbors many different variants like Barth syndrome, a new gene editing therapy must be designed for each person’s specific variant which poses extra expenses and regulatory hurdles for drug developers. One issue that both gene addition and gene editing therapies currently struggle with is reaching a broad variety of cell types in the body without causing issues like toxicity. In KJ’s case, the primary target was the liver, which an easier target. In Barth syndrome, the therapy must hit the heart and skeletal muscle effectively without causing side effects. Gene editing therapies may be associated with less side effects but, in the case of systemic treatment, may also require repeat dosing. Gene addition therapy can be associated with significant immune system side effects and require immunosuppression as part of the treatment regimen but is designed to ideally only be given one time.
Take Home Message
As scientists continue to improve upon each of these therapeutic approaches and as regulatory frameworks adapt to support more of a platform-based approach towards gene therapy, the costs and time needed to develop gene therapies will be reduced. KJ’s case report is a significant step in the right direction towards developing custom treatments for all ultra-rare and rare diseases.


